依据中国的法律体系,我们可以将医疗器械人因工程方面的法律法规划分为三个层面:顶层为法律层面,一般为法律或者条例,以及实施细则等;中间层面就是标准,有国家标准,国家推荐标准,行业标准,行业推荐标准,我们国家很多时候都是转化同期的国际标准;在下一个层面就是实施指南,或者技术指导原则,进一步明确实施标准的办法和要求。
一、法律层面
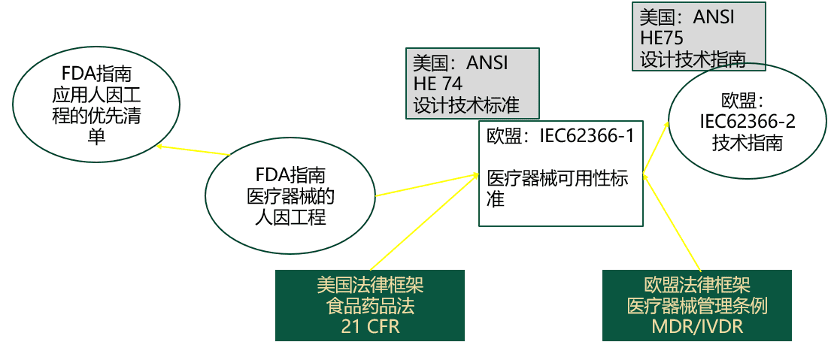
目前,在法律层面,美国和欧盟的立法较早,其中《美国联邦法规》(CFR)中21篇说明了食品与药品的相关要求,也是美国食品药品监督管理局(FDA)管理药品的法律依据。医疗器械在通过美国的审批后会被授予注册证书。
欧盟于2017年5月5日发布了欧盟医疗器械法规(MDR)和体外诊断医疗器械法规(IVDR),得益于完善的法律体系,虽然欧盟没有统一的监督管理局,但是仍然能够通过授权公告机构(NB)发布CE认证来实现对医疗器械上市的监管。
中国于2020年12月21日由国务院修订通过《医疗器械监督管理条例》,类似于美国,该条例是国家药品监督管理局(NMPA)进行药品管理的法律依据。值得注意的是,该条例中并没有关于医疗器械可用性工程的相关条款。
二、标准层面
在医疗器械的质量管理标准层面,美国主要根据FDA质量管理规范(QSR)即CFR21.820,规定了在设计、制造、包装、标签、贮存、安装和服务中的要求,核心是围绕医疗器械本身的安全性和有效性。欧盟方面的质量管理标准依据ISO 13485:2016质量管理体系(QMS)-医疗器械;中国方面则根据国际标准ISO 13485做了相应的修订,对应的行业标准为YY/T0287和YY/T0288,其中YY/T0287已于2022年10月12日升级为国家标准GB/T42061,将于2023年11月1日起正式实施。
在为质量管理服务的风险管理标准上,美国及欧盟都认可ISO 14971医疗器械风险管理,这份文档说明了医疗器械风险管理的术语,原则和流程,包括软件型医疗器械和体外诊断医疗器械。中国方面则根据ISO 14971-2005修订了YY/T 0316-2016作为医疗器械风险管理的标准。该行业标准也于2022年10月12日升级为国家标准GB/T42062-2022,将于2023年11月1日起正式实施。
医疗器械可用性工程作为风险管理过程的一部分,也有相应的管理标准。美国方面采用HFE共识标准中的ANSI/AAMI HE74:2001 (R2009),该文档对标国际标准IEC62366-1,提供了结构化的方法帮助生产商开发安全和有效的医疗设备。欧盟依据IEC62366-1 医疗器械可用性标准进行管理,该标准主要关注可用性工程作为医疗设备用户界面的设计和开发过程,以识别和减少使用错误的可能性和使用相关的风险。中国则根据IEC62366-1修订了YY/T 1474-2016。目前中国在医疗器械可用性方面采用的标准全都是推荐标准而非强制性标准,需要通过行政命令或相应的指导原则才能使相关标准得到进一步的执行。
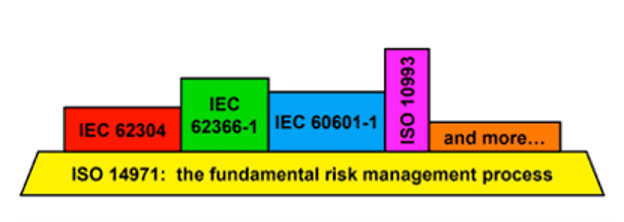
另外,针对医疗器械开发的不同方面,还有不同的补充标准,如针对医疗电气设备可用性安全的EN60601-1-6,中国据此标准修订了GB9706,其中YY/T 9706.106是专门针对可用性的。美国及欧盟采用的IEC62304是针对医疗器械软件生命周期流程的;ISO16142-1-2016是医疗器械安全性和性能的公认基本原则和标准选择指南。
三、操作指南
在法律层面和标准层面想要具体落实可用性工程都有相当的难度,法律和标准主要阐明了应该做的标准,而指南则给出了具体怎样做的结构性方法。美国方面依据FDA医疗器械的人因工程指南和FDA应用人因工程的优先清单指南,另外,还有对标IEC62366-2的ANSI/AAMI HE75:2009 (R2013)HFE共识标准。欧盟方面则依据IEC62366-2技术指南。中国方面目前尚缺少相关指南,但医疗器械人因设计技术审查指导原则(征求意见稿)和家用医疗器械说明书注册技术审查指导原则(征求意见稿)已经完成,正在等待两份文件正式版的发布。这两个审查指导原则的正式发布和实施,以及随之而来的相关行政文件,将使得整个医疗器械可用性方面的法律法规框架更加完善,并推进整个框架的进一步执行。
具体来看,医疗器械人因设计技术审查指导原则(征求意见稿)针对中高风险医疗器械(第二、三类医疗器械)提到了两点之前并没有强制规定的:1、新注册的产品需要提供医疗器械人因设计的研究报告。2、进口医疗器械原则上应在中国开展人因设计确认测试,除非能够提供数据详实的支持材料证实中外差异对于人因设计确认测试无显著影响。
其中,指导原则对医疗器械人因设计研究报告的内容做了具体的说明,报告需要注明产品基本信息、使用风险级别并说明用户,使用场景和用户接口三个核心要素。另外,在设计过程、需求规范和风险管理上应该有详细的说明。最后,指导原则提供了总结性可用性测试和等效医疗器械对比评价报告两种方法,报告需要做到可追溯性分析并给出安全性和有效性结论。这就要求企业建立可用性工程过程和可用性接受准则并最终证明产品能够满足接受准则。
对医疗器械人因工程法律法规不熟悉?
全面了解医疗器械人因工程

